※本資料における条文等の解釈について、厚生労働省の確認を得たものではありません
TRにおけるGMP・GLP・GCPについて
予め必要な基準等を把握し、管理や記録等の手順を適切に定めておくことで、資料作りのための再試験等の必要性が減少する可能性があります。
被験者の方への安全性を、これまで以上に担保できる可能性があります。
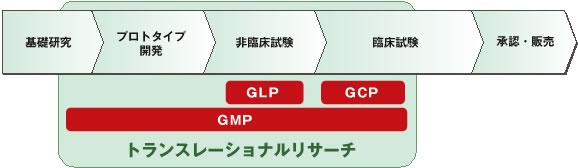
GMP(Good Manufacturing Practice)とは
薬事法第14条第2項に「次の各号のいずれかに該当するときは、前項の承認は、与えない。」とあり、同項第4号に「申請に係る医薬品、医薬部外品、化粧品又は医療機器が政令に定めるものであるときは、その物の製造所における製造管理又は品質管理の方法が、厚生労働省令で定める基準に適合していると認められないとき。」とあります。
この「厚生労働省令で定める基準」として、平成16年12月24日付 厚生労働省令第179号(医薬品及び医薬部外品)と平成16年12月17日付 厚生労働省令第169号(医療機器及び体外診断用医薬品)がそれぞれ告示されています。
二つの省令による基準(省令GMP)は、薬事法の承認要件とされていることから、薬事承認を取得するためには、これらの基準に合致している必要があります。
最終的な目標が薬事承認であったとしても、TRでは承認取得までを範疇にはしておらず、省令GMPに合致しないことをもって、適当な製造管理・品質管理体制ではないとは言えないと思われます。
TRにおけるGMPとは
治験実施に関する基準、いわゆる省令GCPでは、治験薬又は治験機器の品質管理及び製造管理を以下のように規定しています。
医薬品省令GCP第17条
「治験依頼者は、治験薬の品質の確保のために必要な構造設備を備え、かつ、適切な製造管理及び品質管理の方法が採られている製造所において製造された治験薬を実施医療機関に交付しなければならない。」
(第26条の3に、医師主導治験に関する条文があります)。医療機器省令GCP第25条
「治験依頼者は、治験機器の品質の確保のために必要な構造設備を備え、かつ、適切な製造管理及び品質管理の方法が採られている製造所において製造された治験機器を実施医療機関に瑕疵のない状態で交付しなければならない。」
(第36条に、医師主導治験に関する条文があります。)
医薬品の治験においては、「治験薬の製造管理及び品質管理基準及び治験薬の製造施設の構造設備基準(治験薬GMP)について」という通知が発出されています(平成9年3月31日付 薬発第480号)。
TRが、医薬品や医療機器の開発を円滑に進めていくことを目的のひとつとするのであれば、少なくとも治験における品質管理や製造管理の体制を構築しておくことが必要と思われます。もちろん、省令GMPに準拠していることが、望ましいことは言うまでもありません。
GLP(Good Laboratory Practice)とは
GLPとは、医薬品や医療機器の安全性に関する非臨床試験の実施に関する基準と言えます。その趣旨は、以下のように省令に定められております。
医薬品では、
「この省令は,薬事法(昭和35年法律第145号。以下「法」という。)第十四条第三項(同条第六項,法第十九条の二第四項及び第二十三条において準用する場合を含む。以下同じ。)並びに法第十四条の四第四項及び第十四条の五第四項(これらの規定を法第十九条の四及び第二十三条において準用する場合を含む。以下同じ。)に規定する厚生労働大臣の定める基準のうち,医薬品の安全性に関する非臨床試験(薬事法施行規則(昭和36年厚生省令第1号)第十八条の三第一号ニ(第二十六条の三及び第二十七条において準用する場合を含む。)及び第二十一条の三第一項(第二十六条の十三及び第二十七条において準用する場合を含む。)並びに法第十四条の五第三項(法第十九条の四及び第二十三条において準用する場合を含む。以下同じ。)に規定する資料のうち急性毒性,亜急性毒性,慢性毒性,催奇形性その他の毒性に関するものの収集及び作成のために,試験施設において試験系を用いて行われるものに限る。以下単に「試験」という。)に係るものを定めるものとする。」
とあります。
医療機器では、
「この省令は、薬事法(昭和三十五年法律第百四十五号。以下「法」という。)第十四条第三項(同条第九項及び法第十九条の四において準用する場合を含む。以下同じ。)並びに第十四条の四第四項及び第十四条の六第四項(法第十九条の四において準用する場合を含む。以下同じ。)に規定する厚生労働大臣の定める基準のうち、医療機器の安全性に関する非臨床試験(薬事法施行規則(昭和三十六年厚生省令第一号)第四十条第一項第五号ロ及びニ(第百二条において準用する場合を含む。)及び第五十九条第一項(第百十一条において準用する場合を含む。)並びに法第十四条の六第四項(法第十九条の四において準用する場合を含む。以下同じ。)に規定する資料のうち生物学的安全性に関するものの収集及び作成のために、試験施設において試験系を用いて行われるものに限る。以下単に「試験」という。)に係るものを定めるものとする。」
とあります。
したがって、医薬品や医療機器の承認申請資料のうち、いわゆる毒性試験に関する資料を作成するための試験については、省令GLPに準拠する必要があると言えます。
なお、全ての非臨床試験がGLP基準で行われることを否定するものではありません。
GCP(Good Clinical Practice)とは
GCPは、医薬品や医療機器の臨床試験の実施に関する基準として、省令が定められております。薬事法では、「治験」とは、以下のように定められております。
薬事法第2条第6項
「この法律で「治験」とは、第十四条第三項(同条第九項及び第十九条の二第五項において準用する場合を含む。)の規定により提出すべき資料のうち臨床試験の試験成績に関する資料の収集を目的とする試験の実施をいう。」薬事法第14条第3項
「第一項の承認を受けようとする者は、厚生労働省令で定めるところにより、申請書に臨床試験の試験成績に関する資料その他の資料を添付して申請しなければならない。この場合において、当該申請に係る医薬品又は医療機器が厚生労働省令で定める医薬品又は医療機器であるときは、当該資料は、厚生労働大臣の定める基準に従つて収集され、かつ、作成されたものでなければならない。」
従って、承認申請に添付する臨床試験成績を実施する基準として、省令GCPが定められていると言えます。
TRにおいては、必ずしも「治験」を実施するとは限りませんが、臨床試験結果を医薬品等の開発に円滑につなげていくためには、可能な限り省令GCPに準拠した形で臨床試験を実施していくことが望ましいと思われます。

